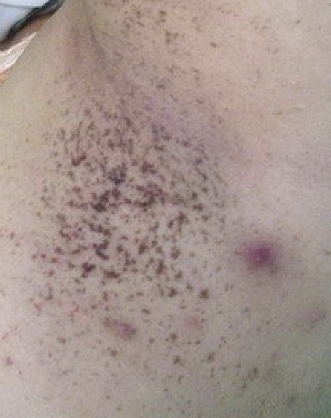
Dowling-Degos disease (also known as reticulated pigmented anomaly of the flexures) is an inherited pigmentary dermatosis characterized by progressive reticulated hyperpigmentation of the skin of the flexures and body folds. ICD-10 Code: L81.8
The disease typically occurs between the ages of 20 and 40, less commonly in adolescents, and affects men and women equally. The disease was first described in people from Asian countries, but cases have been reported worldwide. The pattern of inheritance is autosomal dominant.
The disease is thought to be caused by a mutation in the keratin 5 gene (KRT5) located on chromosome 12 in the 12q13.13 region. Mutations in the POFUT1, POGLUT1 genes (encoding O-fucosyltransferase 1 and protein O-glucosyltransferase 1, which partially control the proliferation and differentiation of melanocytes and keratinocytes) and the PSENEN gene (encoding a component of the γ-secretase protein complex) have also been reported in cases associated with hidradenitis. However, there are also observations of sporadic cases of the disease.
The disease begins with the eruption of small 2-5 mm macules that gradually coalesce into reticulated patches with a coalescing hyperpigmented center and isolated macules at the periphery. The color of the rash varies from light yellow-brown to black. Initially, the affected areas are localized to the groin folds, inner thighs, perianal region, and as the disease progresses, the rash spreads to the inframammary folds, breasts, nipples, axillary folds, neck, elbows, knees, and forearms. Rarely, the face and scalp may be affected.
In most cases, there are eruptions of small comedone-like hyperkeratotic follicular papules in addition to reticulated pigmented macules, leaving punctate acneiform scars. These papules are initially localized around the mouth and nose and then spread to the entire face, neck and back.
Other less common manifestations include tricholemmal and epidermoid cysts, soft fibromas, nail dystrophy, generalized hypotrichosis, and developmental delay. Eruptions are usually asymptomatic, but sometimes mild pruritus may be observed.
Clinical variants:
- Variant associated with hidradenitis suppurativa (inverse acne) in the groin folds and axillary folds.
- Acantholytic variant (Galli-Galli disease)
- Follicular variant
- Localized variant
- Hypopigmented variant - appearance of hypopigmented spots alongside classical generalized eruptions.
- Congenital dyskeratosis
- LEOPARD syndrome
- Confluent and reticulated papillomatosis of Gougerot and Carteaud
- Dyschromatosis symmetrica hereditaria of Dohi
- Reticulate acropigmentation of Kitamura
- Granular parakeratosis
- Acanthosis nigricans
- Dermatopathia pigmentosa reticularis
- Neurofibromatosis type 1
- Haber syndrome