Interstitial granulomatous dermatitis is a clinical and histopathologic variant of reactive granulomatous dermatitis associated with various autoimmune diseases, characterized by the appearance of linear and annular plaques. ICD-10 Code: L92.
It typically occurs in people between the ages of 50 and 60, more often in women. The prevalence is unknown and it is considered a rare disease.
The etiopathogenesis is unclear. It is thought that the initiating factor is the deposition of immune complexes on or around small dermal blood vessels due to the underlying disease. This leads to complement and neutrophil activation, resulting in degeneration of dermal collagen and slowed blood flow to the affected area. Collagen degeneration triggers an immune response resulting in a granulomatous palisading lymphohistiocytic infiltrate. A moderate but persistent level of immune complexes may contribute to this phenomenon. Exogenous trauma exacerbates or accelerates the process.
Associated Conditions:
- Rheumatoid arthritis – The combination with it is often referred to as interstitial granulomatous dermatitis with arthritis (Ackerman's syndrome, IGDA).
- Autoimmune diseases (systemic lupus erythematosus, systemic vasculitis, antiphospholipid syndrome, juvenile idiopathic arthritis, Still's disease, Sjögren's syndrome, Churg-Strauss syndrome, Behçet's disease, autoimmune thyroiditis, autoimmune hepatitis)
- Neoplasms (lymphoproliferative disorders, bronchial squamous cell carcinoma)
- Lyme disease (borreliosis)
- Pulmonary silicosis
- Pulmonary paracoccidioidomycosis
- In cases associated with drug use (angiotensin-converting enzyme inhibitors, calcium channel blockers, beta-blockers, lipid-lowering agents, antihistamines, anticonvulsants, antidepressants, tumor necrosis factor-α blockers, and sennoside), the term interstitial granulomatous drug reaction is used.
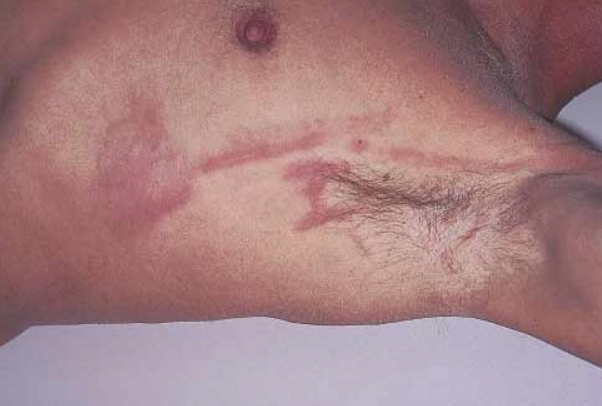
The disease is characterized by the eruption of dense linear subcutaneous stripes, 3-5 mm wide and 10-40 cm long, of pink color, most commonly located on the lateral surface of the trunk from the axillary fold to the iliac crest (the "rope sign" symptom), less frequently on the abdomen, buttocks, inner surfaces of the arms and thighs.
Another characteristic symptom is the presence of circular erythematous plaques with a scaly surface, often coalescing into extensive patches. The eruptions are usually symmetric. Less common manifestations include diffuse maculopapular rash, subcutaneous flesh-colored nodules, large atrophic hyperpigmented plaques, periungual erythema and mucosal involvement, and papules on the elbows.
Typically, the disease progresses asymptomatically, but in cases of arthritis (which may precede, accompany, or follow the eruptions), arthralgia and myalgia may occur. The course of the disease is chronic and recurrent. The duration of the attacks varies from 1 week to several months. Spontaneous resolution occurs in 20% of patients.
- Palisaded neutrophilic granulomatous dermatitis
- Urticarial vasculitis
- Erythema migrans
- Erythema marginatum
- Morphea
- Systemic lupus erythematosus
- Granuloma annulare
- Cutaneous T-cell lymphoma, granulomatous type
- Erythema annulare centrifugum
- Lichen planus
- Lichenoid drug eruption
- Annular elastolytic giant cell granuloma
- Sarcoidosis
- Necrobiosis lipoidica
- Necrobiotic xanthogranuloma
There is no standard treatment. Treatment of the underlying disease or infection may result in significant improvement. The following medications have been used with varying degrees of success:
- Topical and systemic corticosteroids
- Non-steroidal anti-inflammatory drugs
- Colchicine
- Hydroxychloroquine
- Allopurinol
- Cytostatics (methotrexate, azathioprine, cyclosporine)
- Dapsone
- Tumor necrosis factor inhibitors (etanercept, infliximab, ustekinumab)
- Phototherapy