The term "xanthoma" refers to cutaneous lesions of various clinical presentations characterized by the consistent presence of xanthomatous or foam cells. ICD-10 Code: E78.2
Xanthomatous cells consist primarily of macrophages containing sudanophilic material - lipid droplets that form large or small clusters. Different types of xanthomas are distinguished by the nature of the xanthomatous cell clusters, the presence of other cell types, and the chemical composition of the intracellular lipid droplets. In most cases, xanthomas result from hyperlipidemia, either primary (due to a genetic predisposition to lipid disorders) or secondary (due to various diseases unrelated to lipid metabolism).
The exact mechanisms responsible for xanthoma formation are not fully understood. Several hypotheses have been proposed regarding the mechanisms that may lead to the formation of such cutaneous lesions. These include local inflammation, tissue injury, the presence of low-density lipoprotein (LDL) acetyl receptors on skin macrophages, and increased in situ lipid formation. Since dermatologists are often the first to encounter these cutaneous manifestations, it is necessary to determine the prognostic significance of each type of xanthoma in order to recommend appropriate treatment. It is important to note that in addition to xanthelasma, other forms of xanthoma have been thought to occur in individuals with normal cholesterol levels. In most cases, however, serious metabolic abnormalities were found after thorough lipid and lipoprotein analysis.
Family history should also be taken into consideration seriously. Furthermore, the presence of xanthelasma in patients with normal cholesterol levels does not exclude the risk of atherosclerosis.
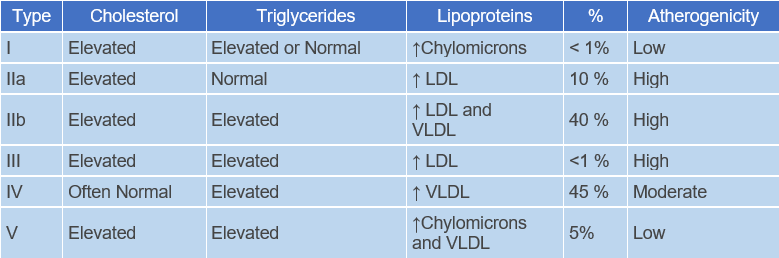
Classification of Hereditary Hyperlipoproteinemias
- Familial Hypercholesterolemia - (Type IIa)
- Familial Dysbetalipoproteinemia - (Type III)
- Familial Combined Hyperlipoproteinemia - (Type IIa, IIb, IV)
- Familial Hypertriglyceridemia - (Type IV, V)
- Familial Lipoprotein Lipase Deficiency - (Type I)
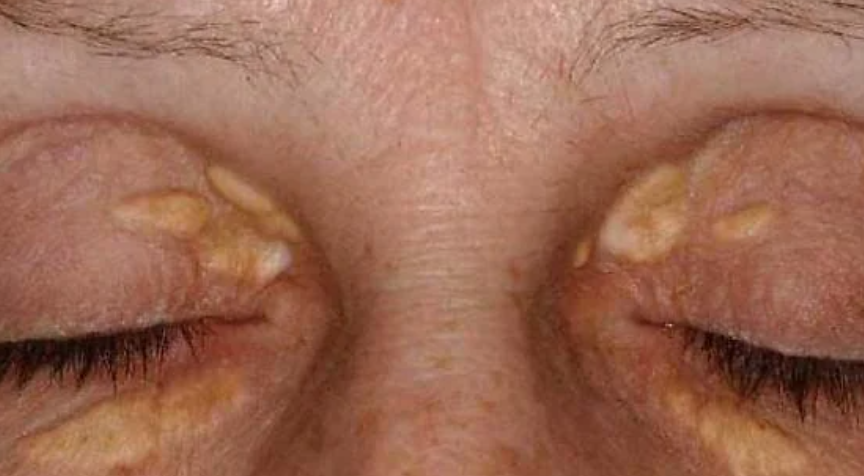
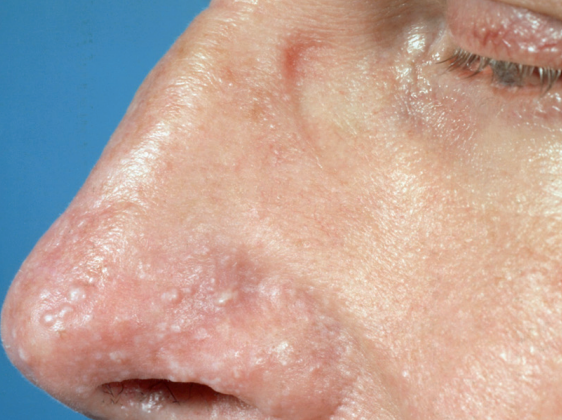
Xanthelasma
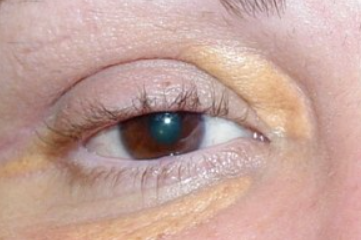
Develops more commonly in older individuals, especially women, on the eyelids and periorbital area as flat, slightly elevated, soft papules or plaques of round, oval, or elongated shape, ranging in diameter from 2-3 to 10 mm or more. They are straw-yellow in color, have a smooth or wrinkled surface, and cause no subjective sensation. Approximately 50% of patients with xanthelasma have normal plasma lipid levels, while the remainder usually have elevated cholesterol levels (this is particularly characteristic of younger patients).
Main Causes:
- Type II hyperlipoproteinemia (familial)
- Type III hyperlipoproteinemia (familial)
- Mixed hyperlipidemia (Type IV)
- Apolipoprotein E phenotypes
- Primary biliary cholangitis
Palmar plane xanthoma

It is characterized by the appearance of yellow-orange flat stripes in the folds of the palms and fingers with clear borders, as well as flat papules of the same color, 4-6 mm in diameter, localized only in the palm area. The condition appears after the age of 30. It can be inherited as an autosomal recessive trait (more common) or as an autosomal dominant trait (less common). It is associated with early atherosclerosis of the peripheral vessels, ischemic heart disease, liver disease, diabetes, often with obliterative atherosclerosis of the arteries of the lower limbs.
Main Causes:
- Homozygous familial hyperlipidemia
- Type III hyperlipoproteinemia (familial)
- Diabetic Xanthomatosis
- Prolonged cholestasis
- Monoclonal gammopathy
Generalized (Diffuse) Xanthoma
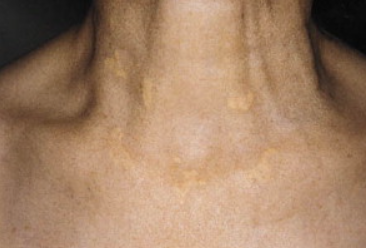
It is characterized by flat, oval, or linear yellow plaques that are more elevated above the skin surface than xanthelasma. These plaques may start as localized lesions but gradually, due to slow progression, spread to the skin of the face, neck, upper trunk, upper limbs and folds.
It may develop either for no apparent reason in cases of normolipidemia or in patients with multiple myeloma (sometimes long before its clinical manifestation), paraproteinemia, as well as acquired deficiency of C1 esterase inhibitor.
Main Causes:
- Primary hyperlipidemia type III
- Secondary hyperlipidemia
- Monoclonal gammopathy associated with myeloma, lymphoma, leukemia, rheumatoid arthritis
- IgA gammopathy with hypolipidemia
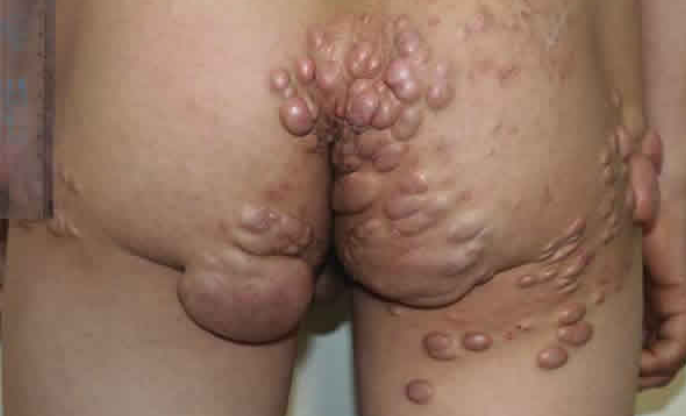
Tuberous Xanthoma
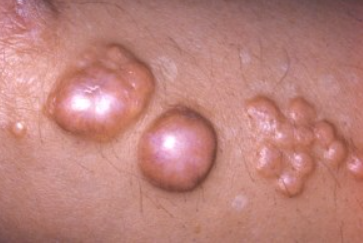
It begins with the appearance of small, soft, yellow, red, or flesh-colored papules on the extensor surfaces of the knees, elbows, shoulders, and buttocks. Over time, these become large (up to 1-5 cm) nodules with a denser consistency due to fibrous tissue infiltration. They may be yellow, orange, or purplish red in color and often coalesce into massive conglomerates. Some elements may be surrounded by a reddish-blue rim.
There is an elevated plasma cholesterol level with an unchanged ratio of free and esterified fractions (there are cases of normocholesterolemia), rarely triglycerides. Patients often have a very high frequency of vascular atherosclerosis. Cases of transformation of xanthomas into keloid scars have been described. Mucous membranes may be involved. Early involvement of blood vessels, joints, and bones is noted. The liver and spleen are often involved.
Main Causes:
- Hyperlipoproteinemia type II (familial)
- Hyperlipoproteinemia type III (familial)
- Secondary hyperlipidemia, associated with, hypothyroidism, primary biliary cholangitis, nephrotic syndrome
- Monoclonal gammopathy
- Xanthomatosis ceribrotendinosus
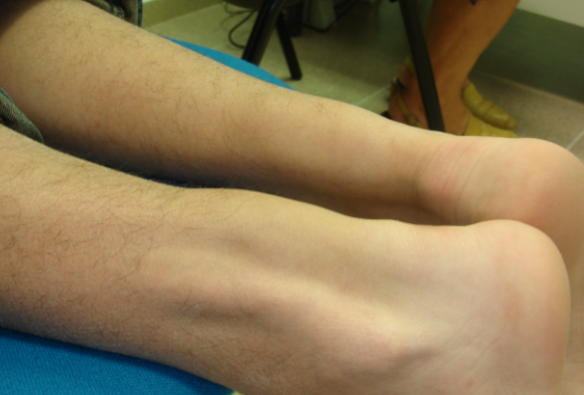
Tendinous Xanthoma
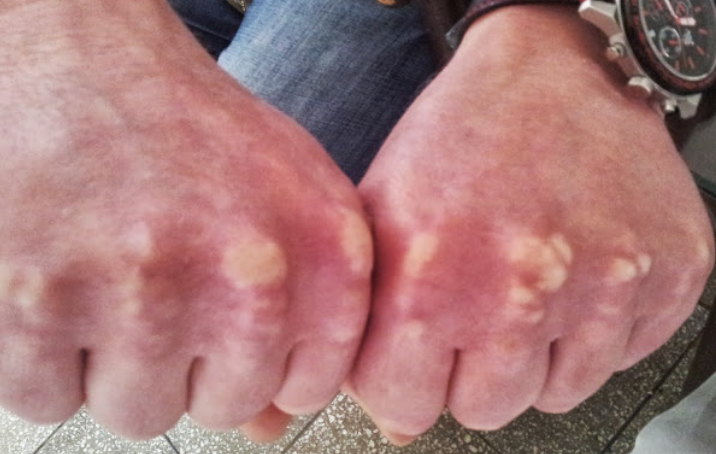
Classically, tendinous xanthomas are dense, smooth, deep-seated, slowly growing tumor-like formations within the tendons of the wrist extensor, knee, and Achilles tendons. The latter may cause pain and tenderness to palpation due to an inflammatory reaction. The skin overlying the xanthoma remains unchanged and is not adherent to the xanthoma. Trauma is considered a predisposing factor for their formation.
Main Causes:
- Primary systemic hyperlipidemia
- Hyperlipoproteinemia type II (familial)
- Apolipoprotein E phenotypes
- Secondary hyperlipidemia with prolonged cholestasis
- Xanthomatosis ceribrotendinosus
Eruptive Xanthoma
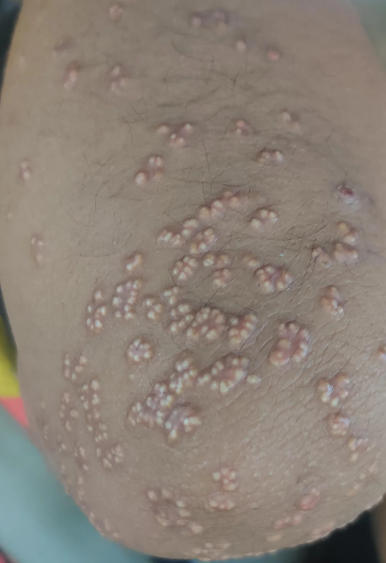
It occurs exclusively in association with elevated plasma triglyceride levels, often in cases of secondary hypertriglyceridemia caused by uncontrolled diabetes. The eruptions occur suddenly, typically in clusters, and, unlike other forms of xanthomatosis, may be accompanied by pruritus.
Papules are small (1-4 mm in diameter), soft, flat or (more commonly) hemispherical, yellowish or yellow-orange in color, and surrounded by an erythematous halo. They are most commonly localized over areas subjected to pressure and on the extensor surfaces of the limbs (joint areas, buttocks), sometimes widespread, including mucous membranes. Perifollicular and follicular xanthomas with cystic changes have been described.
Main Causes:
- Hyperlipoproteinemia type I (familial lipoprotein lipase deficiency)
- Hyperlipoproteinemia type III (familial)
- Hyperlipoproteinemia type IV (familial hypertriglyceridemia)
- Hypertriglyceridemia type V
- Secondary hyperlipidemia associated with diabetes mellitus, alcoholism, estrogens and contraceptives, retinoids, hyperthyroidism, nephrotic syndrome
Perineural Xanthoma
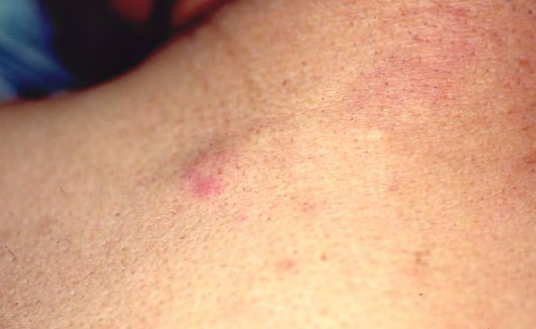
Verruciform Xanthoma
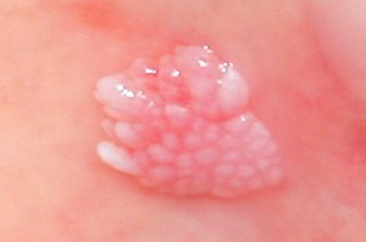
Xanthoma disseminatum
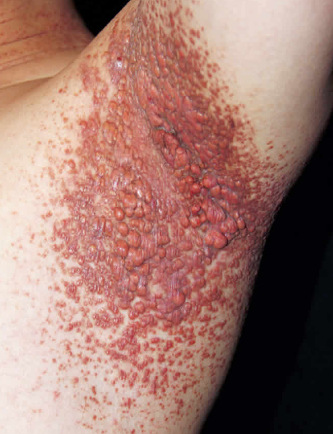
Also known as Montgomery Syndrome. Some authors classify this condition as normolipidemic xanthomatosis, while others associate it with the group of non-Langerhans cell histiocytoses. The eruptions appear as reddish-brown papules 2-10 mm in diameter that rapidly evolve into larger yellow-brown confluent patches resembling eruptive xanthomas. They are typically clustered and localized on the proximal parts of the limbs, flexures, trunk, and face. Dark brown plaques may form on the flexural areas of adults.
Mucous membranes of the mouth, larynx, trachea, conjunctiva, cornea, and sclera are often involved. Respiratory lesions may cause hoarseness and shortness of breath.
The prognosis is generally good. The disease resolves spontaneously with involution and scarring of the xanthomas. However, there are cases of fatal outcome due to airway obstruction by xanthomas and development of xanthomatous tumors in brain tissue.
Papular Xanthoma
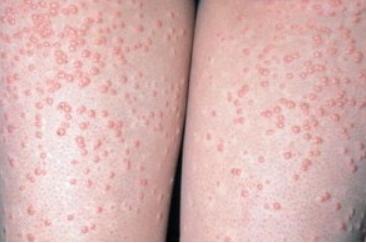
Papular xanthoma is a normolipemic histiocytosis not associated with Langerhans cells. It is observed between the ages of 13 and 57 years with a male to female ratio. Clinically, it manifests as single or multiple yellow-red or yellow-brown papules on the skin of the face, trunk, and extremities. In adults, the mucous membranes may also be involved.
In children, the disease usually resolves on its own, leaving a scar-like appearance, but in adults the disease may progress. There is no systemic involvement. Serum lipid levels remain within normal limits, although qualitative abnormalities of lipoproteins may be present.
Familial Lipoprotein Lipase Deficiency
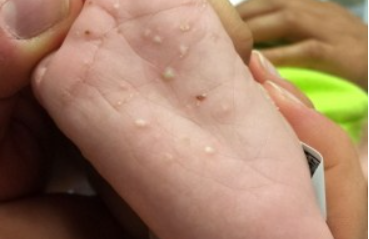
Also known as familial hyperlipoproteinemia or familial chylomicronemia. It is an inherited disorder of lipid metabolism characterized by xanthomatosis and neurological and cardiovascular symptoms.
Characteristic features include multiple tuberous xanthomas around the elbows and knees and eruptive xanthomas on the trunk, eyelids, palms, and soles. The skin is yellowish or golden in color. The disease is associated with hepatosplenomegaly, fever, and involvement of the nervous and cardiovascular systems.
There are six types of familial hyperlipoproteinemia: lipoprotein deficiency due to lipase deficiency (gene 8p22); hyperbeta lipoproteinemia (gene 19qB2); lipoidosis with low-density lipoproteins (gene 19q13. 2); lipoidosis with high levels of very-low-density lipoproteins (gene 11q23); lipoidosis combined with high levels of triglycerides and glucose (gene 11q23); and combined lipoidosis characterized by elevated levels of triglycerides and cholesterol. The inheritance of the pathological gene can be autosomal dominant (types 2, 3, 4, and 6) or autosomal recessive (types 1 and 5).
Dermatochondrocorneal dystrophy
Sitosterolemia
Also known as phytosterolemia or β-sitosterolemia. It's a lipid metabolism disorder characterized by tendon and nodule xanthomas and a predisposition to premature coronary atherosclerosis. Xanthomas of the tendon, earlobes, extensor musculature, and tendons are observed from early childhood. In the elbow area they take on a nodular character.
Blood tests show significantly elevated levels of beta-sitosterol, while cholesterol levels are normal. It's thought that the absorption of β-sitosterol in the intestine is disturbed. Inheritance of the disease gene is autosomal recessive.
Cerebrotendinous Xanthomatosis
It's a normolipidemic xanthomatosis characterized by xanthomas of the tendons, lungs, and central nervous system. This autosomal recessive disorder is characterized by increased synthesis of bile acids, disruption of cholesterol metabolism, and deposition in various organs.
The disease begins with generalized symptoms. Intellectual disability develops in early childhood and progresses each year. Ataxia, spastic paralysis, and cataracts develop between the ages of 12 and 15. Later, xanthomas appear around the Achilles and other tendons. Early atherosclerosis often develops.
Blood cholesterol levels remain unchanged, while cholestanol levels are significantly elevated. The prognosis for recovery and life is poor; patients often die before the age of 20.
Diagnosis is based on the characteristic clinical picture, histological findings and lipid profile blood test.
Xanthelasma
- Epidermal cyst
- Basal cell carcinoma
- Hydrocystoma
- Molluscum contagiosum
- Milia
- Necrobiotic xanthogranuloma
- Sebaceous gland hyperplasia
- Lipoid proteinosis
- Syringoma
- Erdheim-Chester disease
Palmar plane Xanthoma
- Aquagenic palmoplantar keratoderma
- Palmar fibromatosis
Tendinous xanthoma
- Giant cell tumor of the tendon sheath
- Rheumatoid nodules
- Subcutaneous granuloma annulare
- Knuckle pads
- Erythema elevatum diutinum
Tuberous Xanthoma
- Erythema elevatum diutinum
- Multicentric reticulohistiocytosis
Eruptive xanthoma
- Disseminated granuloma annulare
- Bacterial Folliculitis
- Pityrosporum folliculitis
- Xanthomatous variant of necrobiosis lipoidica
- Xanthelasmoid mastocytosis
- Disseminated juvenile xanthogranuloma
- Multicentric reticulohistiocytosis
- Steroid acne
- Sarcoidosis
- Cutaneous leukemia
- Langerhans cell histiocytosis X
- Lichen amyloidosis
Local Treatment
- Mohs surgery
- Cauterization
- Carbon dioxide laser
- Argon laser
- Carbon dioxide laser with ultrapulsation
- Pulsed dye lasers
- Pulsed Erbium:YAG laser
Diet
This therapeutic approach is quite effective when initiated early in life. It has been shown that dietary therapy in 2-7 year old children heterozygous for familial hyperlipidemia can result in a widespread reduction in total cholesterol and LDL levels. The diet should include foods rich in polyunsaturated fatty acids and exclude cholesterol-containing lipids. Olive oil- and legume-rich foods (Mediterranean diet) are an alternative to long-term low-cholesterol diets. However, some children may require additional medication to achieve satisfactory lipid levels. Adults with hypercholesterolemia should also follow such a diet, although it's not the only therapeutic measure. Various types of diets have been tested to achieve optimal metabolic balance, and it has been shown that a diet high in low-calorie carbohydrates and low in fat (90.5% oligosaccharides of glucose, 1.3% vegetable oil, and 8.2% crystalline amino acids) can normalize blood lipid levels and alleviate cutaneous xanthomas in patients with homozygous familial hyperlipidemia.
Pharmacological Therapy
- Bile acid sequestrants (cholestyramine and colestipol) have no systemic effects. They essentially bind bile acids in the intestine, interfering with their enterohepatic circulation and thereby increasing fecal steroid excretion. It should be noted, however, that in some cases these drugs can lead to increased production of very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL) in the liver, exacerbating hyperlipidemia rather than reducing it. Although cholestyramine is effective in reducing LDL cholesterol levels in children with inherited hyperlipidemia, most children cannot continue treatment because of the drug's side effects, including constipation, unpleasant taste, nausea, and flatulence, which is most common. Folate deficiency may also occur and vitamin D supplements are recommended.
- The effect of hypocholesterolemia with nicotinic acid intake is probably mediated by reducing the synthesis of LDL or VLDL in the liver, which is well documented. However, the enhancement of this effect with high doses of nicotinic acid must be tempered by the increased incidence of side effects that may lead to drug intolerance. Hyperemia, nausea, discomfort, dry skin, and in some cases, visual disturbances may occur. Contraindications to prescribing nicotinic acid include active liver disease, hyperuricemia, diabetes, and peptic ulcer. It should be noted that nicotinic acid and bile acid sequestrants, although an acceptable solution for patients with other causes of primary hypercholesterolemia, are not able to control hyperlipidemia and xanthomas in patients with heterozygous familial hyperlipidemia.
- Lovastatin is a well tolerated drug. Common adverse reactions include headache, nausea, fatigue, insomnia, rash, gastrointestinal upset, and in extremely rare cases, myositis. Lovastatin is not indicated for use in children and should be used cautiously in women of childbearing age. Good results have been observed with the combination of lovastatin and LDL apheresis. In some particularly severe cases, the drug has been combined with liver transplantation.
- Other drugs belong to the class of statins (HMG-CoA reductase inhibitors). Simvastatin and pravastatin are the most commonly prescribed. Like lovastatin, simvastatin is prescribed in combination with LDL apheresis. In some cases, simvastatin monotherapy did not prevent the development of peripheral artery disease, but when combined with LDL apheresis, a significant reduction in carotid intima-media thickness was observed without an increase in hemodynamically significant lower extremity stenosis.
- Fibrinic acid derivatives are also effective in hyperlipidemia and may be used as first-line agents. Clofibrate, gemfibrozil, bezafibrate, fenofibrate, and ciprofibrate are the most commonly used. The latter three have the most potent hypocholesterolemic effects. Their mechanisms include increasing lipoprotein lipase activity, increasing LDL catabolism, and reducing VLDL synthesis. These drugs may be prescribed for children. The combination of sitosterol with bezafibrate is acceptable, safe and effective in children at high risk of familial hypercholesterolemia.
- Probucol, although moderately effective in primary hypercholesterolemia, inhibits cholesterol synthesis and transport in human tissues, which explains the observed beneficial effects of this drug on cutaneous xanthomas. Probucol induces regression of tendon xanthomas and xanthelasma. Typically, many patients, especially those with heterozygous familial hyperlipidemia, are treated with two or more lipid-lowering agents with different mechanisms of action that complement or enhance each other's effects.