Xeroderma pigmentosum (XP) - a genodermatosis characterized by increased photosensitivity, skin pigmentation and atrophy, photophobia, neurological symptoms, and a progressive course with a high risk of skin tumors. ICD-10 Code: Q82.1
The disease usually develops in newborns or in early childhood, rarely in the second decade of life. The prevalence in the population of European descent is 2.3 cases per 1 million newborns and the overall prevalence is 4 per 1,000,000. The inheritance pattern is autosomal recessive.
The skin is clear at birth. The first symptoms appear at 6 months of age or later. The prognosis for xeroderma pigmentosum is poor due to the development of malignant tumors with a rapidly progressive course, metastases, and fatal outcome in many patients between the ages of 10 and 15. The risk of developing cutaneous malignancies is hundreds of times higher than in the general population.
In the absence of sun protection, basal cell or squamous cell carcinoma typically develops by 8-9 years of age. The risk of developing melanoma is significantly increased, as is the risk of developing internal organ neoplasms (lung, breast, pancreas, stomach, brain) and leukemia.
Xeroderma pigmentosum is conventionally divided into five stages:
- Erythematous stage: Develops during the first year of life. After brief exposure to sunlight on exposed areas, erythematous-squamous edematous patches appear, often with vesicles or blisters.
- Hyperpigmentation stage: Characterized by the formation of multiple pigmented spots resembling freckles and lentigines that vary in shape, size and intensity of color (from light to dark brown).
- Atrophy stage: In this stage, the skin becomes thinner, dry and tight, with smooth atrophic scars accompanied by telangiectasias, hypo- and/or hyperpigmentation (poikiloderma). Atrophic changes with areas of sclerosis lead to the development of microstomia, ectropion, narrowing of the nasal openings, thinning of the helix and tip of the ear.
- Benign tumor stage: Characterized by the appearance of angiomas, fibromas, papillomas, keratoacanthomas, and actinic keratoses. This can be observed in early childhood (2-5 years).
- Malignant stage: Occurs 8-10 years after the onset of the disease as a progression of previous benign neoplasms or the development of melanoma, sarcoma, squamous cell carcinoma, and basal cell carcinoma. Occasionally, malignant tumors may appear very early, even in the first years of life.
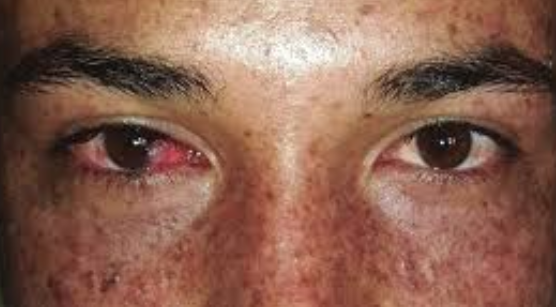
Eye involvement: Ocular pathology occurs in 40% of patients. Most patients present with photophobia (which may be present from the onset of the disease), conjunctivitis (which may be the earliest manifestation), keratitis, blepharitis, iris hyperpigmentation, telangiectasias and pigmentation of the eyelids and conjunctiva, and decreased visual acuity. Later symblepharon, corneal opacity, and growth of benign and malignant conjunctival tumors may develop.
- Bloom syndrome
- Rothmund-Thomson syndrome
- Cockayne syndrome
- Erythropoietic protoporphyria
- Hereditary erythropoietic porphyria
There is no specific treatment. Sun exposure must be strictly limited, and physical and chemical sunscreens, antioxidants, and interferon must be used throughout the day. Early excision of tumors is essential. Prophylactic use of vitamin A and systemic retinoids is important. Imiquimod, fluorouracil and photodynamic therapy are used to treat actinic keratosis and early stages of non-melanoma skin cancer. Regular screening examinations every 3 months are essential for early detection of tumors. Standard methods are used to treat neoplasms.