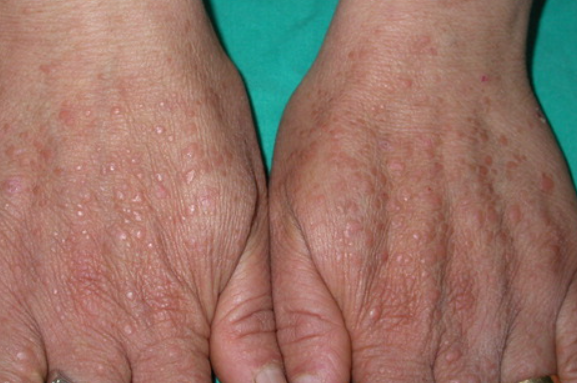
Acrokeratosis verruciformis of Hopf is a genodermatosis classified as a benign neoplasm of the epidermis characterized by verrucous keratotic eruptions on the upper and lower extremities. Its ICD-10 code is Q82.8.
The condition typically develops in early childhood, but in some cases it may be present from birth and persist throughout life. Late onset (sporadic cases) in the third or fourth decade of life is also possible. Females are predominantly affected. The inheritance pattern is autosomal dominant, although sporadic cases have been observed. The etiology and pathogenesis are not fully understood. Some dermatologists consider acrokeratosis to be an "abortive" form of Darier disease or variants of verruciform epidermodysplasia of Lewandowsky-Lutz or epidermal nevus. Recent studies suggest a genetic heterogeneity of the dermatosis, with mutations in the ATP2A2 gene associated with verruciform acrokeratosis and Darier disease. Metabolic disorders leading to vitamin A imbalance are thought to play a role.The eruptions are characterized by dense, hyperkeratotic, hemispherical papules of brownish color measuring 0.5-0.7 cm in diameter and 3-4 mm in height. They may be isolated, grouped, or linearly arranged. Smaller elements (1-2 mm in diameter) have a hemispherical shape and smooth surface.
The distal parts of the limbs, especially the hands and feet, are predominantly affected. The eruptions are located on the dorsal surfaces, less commonly on the palms and soles. Manifestations of the disease may also occur on adjacent skin areas such as the forearms, shins, along the course of the radial and median nerves, and the Achilles tendon.
Occasionally, a linear arrangement of the elements and punctate keratosis on the palms and soles may be observed. Leukonychia, nail deformities, subungual hyperkeratosis, and changes in dermatoglyphics are possible. Cases of unilateral localization have been reported.
The disease progresses without subjective sensations and may persist indefinitely. There have been reports of acrokeratosis coexisting with Darier's disease, acanthosis nigricans, and multiple keratoacanthomas.- Epidermodysplasia verruciformis,dysplasia Lewandowsky-Lutz is characterized by disseminated eruptions localized on the trunk and face, as well as dyschromia, which is not typical for acrokeratosis.
- For common warts, such dissemination of eruptions with a tendency to merge, prolonged course, and symmetric involvement are usually not typical. Histological examination reveals parakeratosis and vacuolization of epidermal cells, which are characteristic of common warts, unlike verruciform acrokeratosis.
- In the verrucous variant of Darier's disease, the eruptions may be clinically identical to those in acrokeratosis verruciformis of Hopf, but other manifestations of Darier's disease (such as isolated typical follicular nodules in seborrheic areas, keratoderma, and nail involvement) may still be seen, although sometimes in a milder form. In doubtful cases, histologic examination of the skin is performed, showing dyskeratosis and lacunar formation atypical of acrokeratosis.
- Congenital poikiloderma with hyperkeratosis is inherited in an autosomal recessive trait. It manifests in early childhood with poikiloderma affecting the face, neck, limbs, and buttocks, which is atypical for acrokeratosis. Warty keratosis develops only at the age of 7-10 years (mainly on the bony prominences of the hands, feet, heels, ankles, and knee joints). In addition, microsomia and hypoplasia of bones are characteristic of congenital poikiloderma with warty keratosis.