Cutaneous angiosarcoma, also known as malignant hemangioendothelioma or lymphangiosarcoma, is a malignant tumor that arises from the endothelium of blood and lymphatic vessels. It is coded as C49.9 in the ICD-10 classification.
There are different types of cutaneous angiosarcoma:
- Idiopathic angiosarcoma: It commonly develops in elderly individuals, particularly in males (twice as common as in females). The majority of patients are of European descent. It accounts for about 1% of all sarcomas. The etiology is unknown. In 50% of elderly patients, the tumor arises in areas exposed to ultraviolet radiation, which is considered one of the risk factors. Other factors include certain morphological characteristics of the skin, such as abundant blood supply and a high number of vascular anastomoses in facial and scalp regions. It is possible that these factors, in combination with ultraviolet radiation, contribute to carcinogenesis. Rarely, it may develop at sites of foreign body implantation or recurrent herpetic infection.
- Angiosarcoma associated with lymphedema, also known as Stewart-Treves syndrome or lymphangiosarcoma, occurs in women aged 60-70 years, sometimes in individuals aged 25-35 years, as well as in men and children. It usually occurs 10-20 years after radical mastectomy and radiotherapy, although early onset within 12-24 months has been reported. Radiation therapy is thought to play an important role in the development of lymphedema, as lymphedema is 10 times more common in women who received radiation therapy after mastectomy than in those who underwent radical surgery alone. The tumor may also develop in the presence of chronic acquired or congenital lymphedema, such as Milroy-Meige disease, Maffucci syndrome, lymphedema associated with morbid obesity, or Hodgkin's disease. It should be noted that angiosarcoma associated with developmental vascular anomalies of the skin occur primarily in childhood. Some authors suggest that tumor development is related to angiogenic factors that induce collateral vessel formation in chronic lymphedema.
- Radiation-induced angiosarcoma is observed in 0.05-0.1% of patients who received radiation therapy for various malignancies (such as cervical cancer, endometrial cancer, male genital organ cancer, Hodgkin's disease, etc.), and less frequently after radiation therapy for benign tumors. The latent period between radiation therapy and the development of the tumor ranges from 2 to 50 years. It occurs between the ages of 37 and 81, with an average age of 63. Women are twice as likely to develop the condition compared to men.
- Primary angiosarcoma of the breast exclusively develops in young women aged 25-40.
Idiopathic angiosarcoma
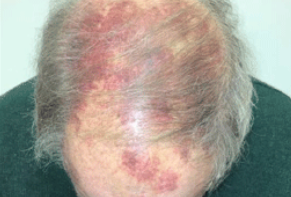
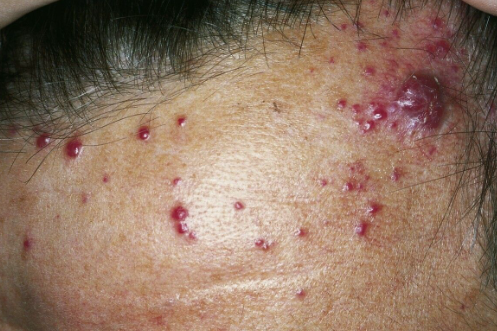
- Superficial spreading type is characterized by rapid centrifugal growth of erythematous patches (often with a brown center and red periphery). Telangiectasias are typically present on the surface. Initial manifestations may resemble bruises with patchy or papular appearance on a background of diffuse edema. Subsequently, soft or firm tumors develop in the form of flat plaques, either singly or in multiple irregularly shaped lesions. They have a deep red color with bluish or purplish tinges and can reach a diameter of 10-15 cm in some cases.
- Nodular type is characterized by the eruption of purplish-brown nodules, sometimes forming clusters of closely packed elements with smooth or verrucous surfaces. Smaller secondary eruptions may be present at the periphery of the main lesions.
- Ulcerative type is characterized by the formation of ulcers with uneven surfaces and serosanguinous crusts.
Localization: Over 50% of tumors arise in the head and neck region (scalp, forehead, temples, cheeks), occasionally involving the lips, nose, and earlobes. Rarely, the oral mucosa may be involved. Other sites of involvement are possible in the remaining cases. Metastases most commonly occur in lymph nodes (approximately 13.5% of cases), and less commonly in the lungs, liver, and bones.
Kasabach-Merritt syndrome (sudden-onset thrombocytopenia) is observed in some patients and may indicate rapid tumor growth or herald the appearance of metastases.
Angiosarcoma associated with lymphedema(Stewart-Treves syndrome)
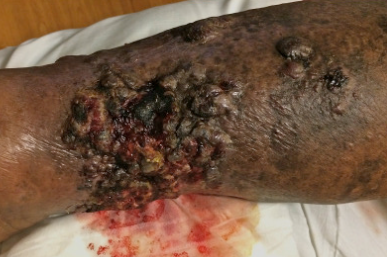
This variant is characterized by the development of multiple bluish-red or dark-red spots in the area of chronic edema, which transform into nodules with a similar or purplish-cyanotic hue. Sometimes, initial manifestations include the formation of blisters. Subsequently, painful nodules, plaques, and conglomerates appear, accompanied by progressive indurated edema. The formation of lymphangio-keratoma, which presents as vesicles with transparent or bloody contents and a pronounced verruciform-hyperkeratotic surface, is also possible.
Tumors tend to quickly ulcerate, spreading both in depth and width. The typical localization is the upper extremities and chest wall. Angiosarcomas not associated with mastectomy can occur on the lower extremities, genitals, and abdominal wall. The disease is aggressive and can be fatal within a few months due to metastasis to internal organs such as the lungs, pleura, liver, and bones.Radiation-induced angiosarcoma
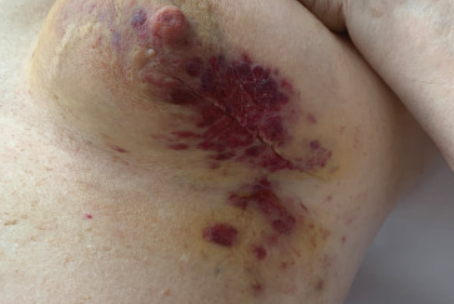
Primary angiosarcoma of the breast
It is characterized by a diffuse erythematous plaque with violet-lilac nodules on the surface in the breast area.
The diagnosis is based on the clinical presentation, medical history, and histological examination.
Immunohistochemical methods are used to determine CD31, Ulex europaeus lectins, and factor VIII. CD31 and factor VIII are considered the most specific markers for angiosarcoma, while the assessment of Ulex europaeus expression is the most sensitive method for diagnosing this type of tumor.- Cellulitis
- Erysipelas
- Breast cancer
- Calciphylaxis
- Squamous cell carcinoma
- Melanoma
- Epithelioid sarcoma
- Pyoderma gangrenosum
- Pyogenic granuloma
- Hematoma / Ecchymosis
- Kaposi's sarcoma
- Intravascular papillary endothelial hyperplasia(IPEH)
- Angiolymphoid hyperplasia with eosinophilia(ALHE)
Due to the tumor's tendency for multifocality and asymptomatic spread, complete surgical removal of the neoplasm is often impossible or ineffective. The prognosis is highly unfavorable, with the average five-year survival rate from the time of tumor detection ranging from 10% to 35% according to different studies. Typical sites of metastasis include the skin, lungs, lymph nodes, spleen, liver, and bones. The presence of metastases is a poor prognostic indicator, usually indicating imminent patient death. Metastases and recurrences of angiosarcoma typically occur within two years of tumor detection. The patient's survival rate is somewhat dependent on the size of the tumor at the time of detection. In general, tumors less than 5 cm in diameter have a more favorable prognosis because they are likely to be recent and have not deeply infiltrated the surrounding tissues. Additionally, a relatively underdeveloped intratumoral vascular network limits the spread of metastases. Other favorable prognostic indicators include a low number of mitoses in the tumor tissue (less than three per high-power field under the microscope), a tumor invasion thickness in the dermis of less than 3 mm, and the absence of recurrence and metastases.
In cases of small-sized angiosarcomas, complete surgical removal of the tumor is possible. To ensure timely detection of recurrences within the first year after detection and excision, patients should undergo medical examinations every 3 months. These exams evaluate lymph node status and may include computed tomography (CT) and/or magnetic resonance imaging (MRI) of the head and neck.