Juvenile xanthogranuloma is observed in young children, mainly under 24 months of age. In nearly 75% of cases, eruptions occur within the first year of life, and in 15% of cases, they are present at birth.
Cases of xanthogranuloma appearing in adults have been described, typically around the age of 20-30, but they can also occur in the elderly.
With solitary eruptions, the condition is slightly more common in boys than girls, with a ratio of 1.5:1. However, in cases of multiple lesions, the predominance of males over females rises sharply to 12:1. The disease is 10 times more common in children of European descent than in other populations.
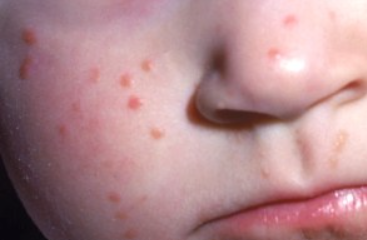
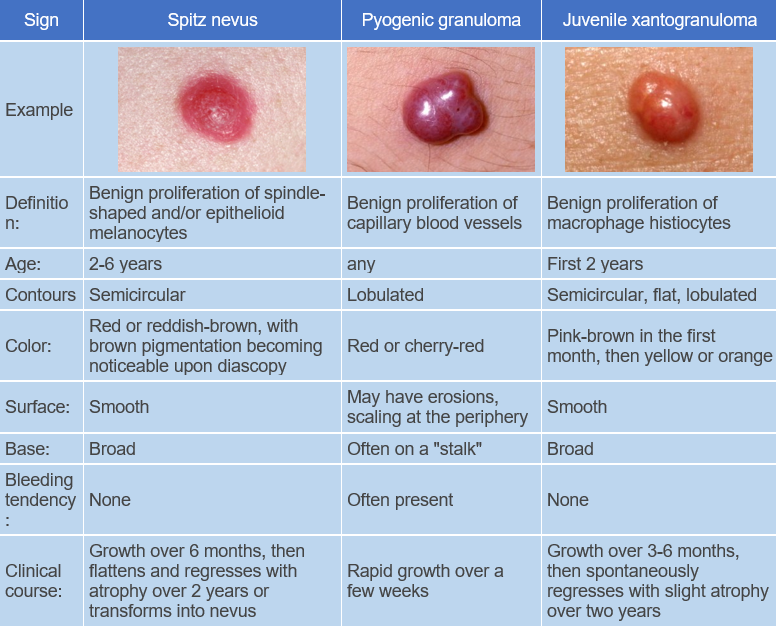
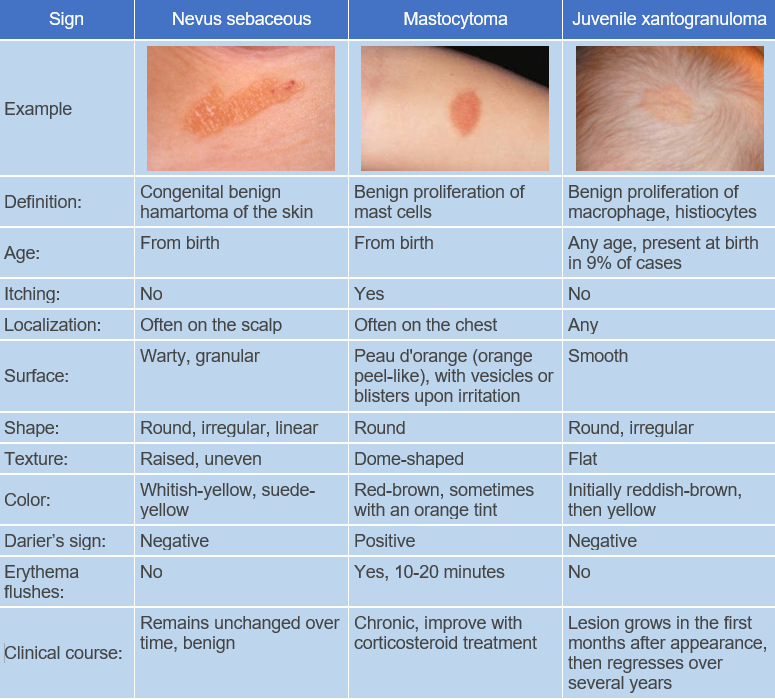
Pathogenesis of the disease is unknown. It is a benign tumor composed of differentiated histiocytes non-Langerhans type. Blood lipids are normal. The reason for progressive lipidization (accumulation of lipids) of histiocytes in the absence of hyperlipidemia is not clear. However, it has been shown that adults with xanthogranuloma have increased uptake of low-density lipoproteins and cholesterol synthesis in macrophages. Some suggest that histiocytes may respond in this way to traumatic or infectious factors.The disease begins with the appearance of pink, reddish-gray macules that rapidly progress to papules, plaques, and nodules. These lesions are moderately firm and elastic to palpation, have well-defined borders, and are often surrounded by a peripheral pink halo. At the onset of the disease, the color of the eruptions ranges from pink to reddish-brown, later becoming pale yellow, orange, and brown with varying shades.
The surface of the lesions is usually smooth, sometimes with prominent skin patterns. Telangiectasias may be observed on the larger formations, and in some cases erosion and ulceration may occur. The most common sites of eruption are scalp, face, neck, upper chest, and less commonly upper and lower extremities and genitalia. Unusual manifestations include eruptions with hyperkeratosis, subcutaneous nodules, and clustered plaques.
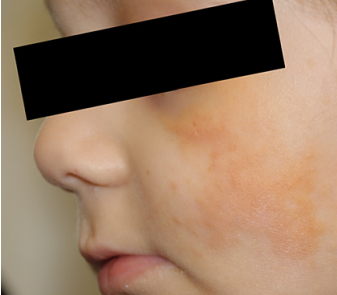
In some patients, "Café au lait" patches may be observed, which may indicate an association with neurofibromatosis.
The disease follows a chronic course, with spontaneous involution of the lesions usually occurring within 1-6 years (average 2-3 years). The involution of the lesions begins with central depression and slight softening, eventually leading to shrinkage and disappearance. Hypo- and/or hyperpigmented patches, mild atrophy, or anetoderma may remain.
Small nodular form
Large nodular form
Plaque form
Giant form
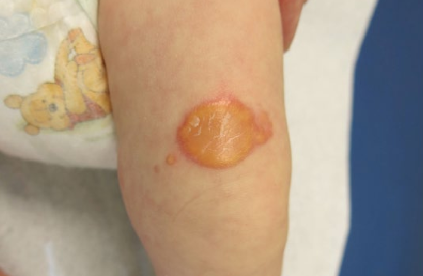
Ulcerative form
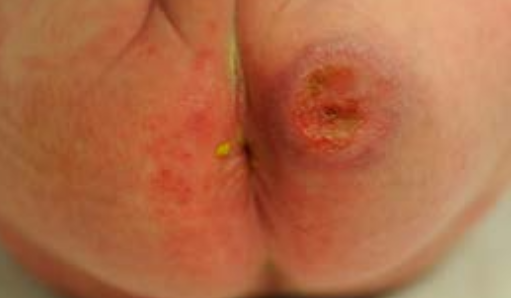
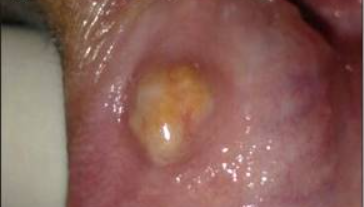
Oral form
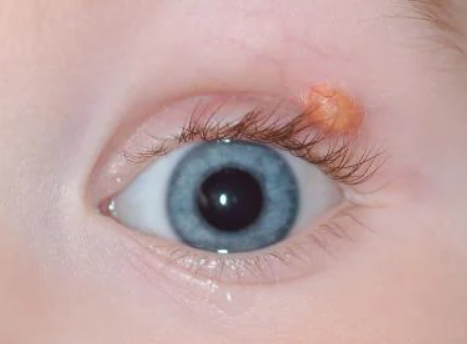
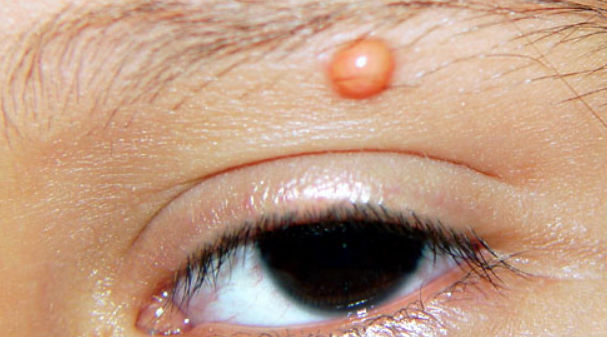
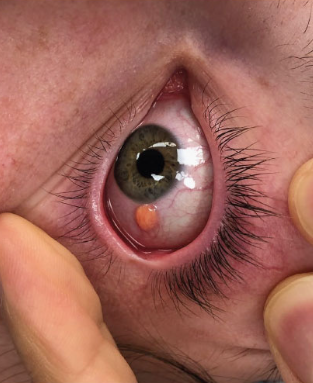
Ocular form
Systemic form
This form occurs in approximately 4% of cases and is associated with cutaneous manifestations in 50% of patients. It is characterized by the formation of granulomas in the muscles, liver, spleen, and lungs. Less commonly, the process may involve bones, testicles and central nervous system, resulting in ataxia, increased intracranial pressure, subdural hemorrhage, developmental delay, non-diabetic diabetes, and other neurologic disorders. Fatal outcomes have been reported in cases of progressive juvenile xanthogranuloma involving the liver in newborns.
The diagnosis is established based on the clinical picture, medical history, and histological examination.
In contrast to X histiocytosis, histiocytes do not tend to penetrate the epidermis, and Langerhans cell granules are not detected by electron microscopy.
Children under 2 years old with multiple eruptions should be under the observation of an ophthalmologist, and those with neurofibromatosis should be under the care of a hematologist.- Mastocytoma
- Spitz Nevus
- Xanthomas
- Langerhans cell histiocytosis.
- Congenital self-healing reticulohistiocytosis
- Melanocytic Nevus
- Dermatofibroma
- Cutaneous reticulohistiocytoma.
- Pyogenic Granuloma.
Patients with cutaneous forms of the disease do not require treatment due to its spontaneous resolution. Surgical excision of lesions may be performed for cosmetic reasons.
In cases of ocular involvement, corticosteroids in the form of eye drops, subconjunctival injections, or oral administration are recommended. Surgery, chemotherapy, or low-dose radiation (300-400 cGy) are used to treat hyphema or glaucoma.
For systemic form, chemotherapy (cytarabine, vincristine), methotrexate, cyclosporine, and high-dose prednisolone are used. Successful use of cladribine has been reported for CNS involvement.